Compressed gases in direct use in the flow of medicinal materials and in process control require a high level of attention and consistent integration into a Good Manufacturing Practices (1,2) system of cleanroom technology. If the supply of compressed nitrogen from a storage facility (gaseous in pressure vessels or as liquid nitrogen with subsequent evaporation) is still qualitatively secured by a Certificate of Analysis (CoA) from the nitrogen manufacturer, it becomes more difficult for the compressed air produced on site: The compression takes place using the ambient air as a resource, which can be very different in various locations or can be strongly affected by environmental influences which have a direct impact on the quality.
Only a purification in the further distribution can produce a suitable quality. To master these general conditions, a specification that is carefully related to the application, a valid processing and distribution technology and, above all, a usage-oriented monitoring of the specifications by monitoring with subsequent approval for use on or in the product is required.
This article is intended to show the relationships between risks and specifications, opportunities and responsibility in validation, and in particular, the use of modern and calibrated measurement technology in the sample chain.
Compressed Air in the Pharmaceutical Environment
Compressed air is an "expensive substance," not only when you look at the costs of using energy to operate a compressed air network in a reliable manner. Valuable also because compressed air often comes much closer to (or in) the product than expected.
In particular, this includes processes such as blowing out primary packaging, transporting products from the container to the filling needle under aseptic conditions, to drying containers, or venting after high-vacuum in lyophilizers or fermenters. Compressed air has an extremely narrow influence on the product and therefore requires a very high level of attention in the GMP (1,2) system.

Shown is an automated filling station in a pharmaceutical plant.
In numerous reports on official inspections, it was often criticized that neither a clear specification, nor a sustainable qualification, or GMP management around the operation of compressed gases was carried out. In particular, the creation of a specification was not carried out or was carried out only inadequately.
The reason for this is certainly that, in contrast to the liquid media, such as aqua purificata or water for injections, there is no clear specification by the European Pharmacopoeia (Ph. Eur.). The specification, ''Air Medicinalis,'' which is found in the Ph. Eur., is unsuitable for the specification of compressed air since it describes breathing air supplied to a patient with acceptance criteria.
The reference to ISO 8573 (3) from the International Organization for Standardization is then only of limited use because here a classification of limit values for particle concentration, pressure dew point and oil content is defined, but it is not recommended in which pharmaceutical application and which class or specification is necessary and shall be used. ISO 8573 also says nothing about possible specifications for airborne germs analogous to Annex 1 of the European Union's GMP guidelines.
This fact has been taken into account as a trend in the recent months in the course of official inspections. The compressed gases nitrogen and compressed air have therefore become the focus of surveillance. Much more: The official opinion on the assessment of GMP obligations for compressed air systems was published in the "Aide memoire - Monitoring of Sterile Manufacturers" of the Central Office of the Federal States for Health Protection in Germany(4), which states:
"When specifying compressed air that comes into contact with the product or surfaces in contact with the product, the following must be observed: In addition to the type of products manufactured, the risk assessment must also take the system design and the quality of the outlet air into account.”
In connection with the evaluation of hydrocarbons, it should be noted that contamination with oil within compressed air is a mixture of oil aerosols, oil vapor and other hydrocarbons. A definition of "oil" as a mixture of hydrocarbons with ≥ 6 C atoms (ISO 8573-1:2010) is therefore appropriate. Measuring methods and recorded oil components must therefore be clarified. Hydrocarbon monitoring is also necessary for oil-free air compressors, since the corresponding contaminants are also introduced via the intake air. The below purity parameters as described in ISO 8573 need to be considered:
- Limits for bacterial count / particles are expected.
- Online monitoring of water and hydrocarbons may be necessary, especially for systems that, due to the use of refrigeration dryers or air compressors with oil cooling, have a higher risk of non-compliance with the specification requirements.
With this clarification there are no limit values according to ISO 8573 as defined, but the direction is clear. In order to define the specification now it is advisable to make a classification based on the Association of German Machine and Plant Manufacturers (VDMA) depending on the application (criticality for the product). As an example, when in use in the sterile area, "Direct contact of the compressed air with the material of a sterile packaging (process air)," is defined in the VDMA 15390 (5) standard sheet with the following acceptance criteria:
- Maximum particle size and density of solid contaminants: Class 1, corresponds to 0.1 µm and 0.1mg/m³.
- Pressure dew point at ambient temperatures> + 10°C: Class 4, corresponds to + 3°C Td.
- Pressure dew point at ambient temperatures <+ 10 ° C: Class 2/3, corresponds to -40°C Td /-20°C Td.
- Maximum oil content: Class 1, corresponds to 0.01 mg / m³.
- Sterility: Yes
Safe Quality Food Standard: 5 Compressed Air Criteria - Webinar RecordingDownload the slides and watch the recording of the FREE webcast to learn:
|
Contamination Risks in Compressed Air
As the main argument for safety in the use of compressed air in a GMP environment, it is repeatedly shown in inspection situations by auditors that the use "oil-free air compressors" are sufficient to ensure qualified and contamination-free compressed air. Unfortunately, this is a fallacy because the risk of contamination is often considerably greater than expected or known. Simplified, it can be divided into two areas:
- Active contamination in compressed air treatment.
- Passive contamination of compressed air by connected air consumer systems.
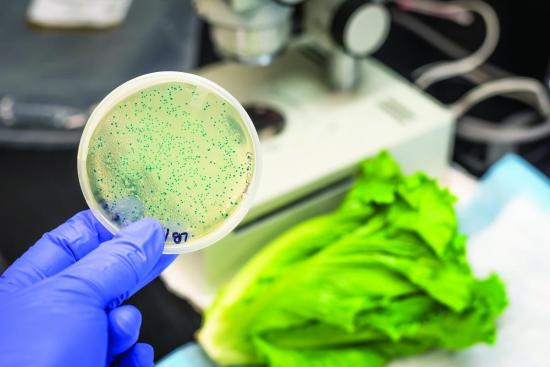
Bacteria culture development in a medium plate.
In the case of active contamination, the primary cause of contamination is found in the use of air compressors with oil cooling. But beyond that, one of the other contaminants needs to be searched for in the aspirated ambient air.
Depending on the position of the openings of the air intake, a considerable amount of particles and oil contaminants in the form of aerosols can be drawn in and compressed from the environment. The (mostly minor) contamination in the form of particles and oil content from the moving parts of an air compressor, especially after a long period of operation, must also be taken into account. In addition, a lot of moisture comes from the air and thus a potential entry of airborne germs into the system in operation and thus into the pipeline network.
In the case of passive contamination, the danger to the compressed air quality is that, due to unfavorable simultaneity factors or insufficient pipe dimensions, "large consumers" cause a reversal of the overpressure in the compressed airline system toward a vacuum system.
In the case of sterile ventilation using compressed air, e.g. containers that fall into the vacuum phase after the vapor phase has collapsed, after steam sterilization, or when the high vacuum of lyophilizers breaks when the valve opens to the compressed airline, the pressure conditions in the line system are reversed. The main risk here is that it can happen in “worst case” situations that such a large, temporary negative pressure can arise in the compressed air network that back-contamination into the system can occur via the compressed airline from other operating areas.
When designing compressed air networks, this should be categorically risk-based checked and planned or taken into account in the in-house change control process before connecting new consumers. To avoid such effects, the use of the so-called "block & bleed circuits" and/or the installation of check valves in the compressed airline could be considered. In any case, this fact should also be checked as part of the Installation Qualification (IQ)/ Operational Qualification (OQ) qualification of the network subject to GMP.
Whether active or passive contamination: In the often very widely branched compressed air networks within a pharmaceutical plant, the principle applies: "What is in the network remains in there!"
In the majority of cases, cleaning is not possible, or will be often not intended in the course of airline planning. In order to make the level of contamination within the compressed airline visible/controllable at all, it is therefore recommended that a so-called "inspection pipe" is installed, i.e. a section of approx. 50 - 100 centimeter (cm), which can be removed and inspected using tri-clamp connections as part of the planned maintenance. In extreme cases, the degree of contamination can then lead to a partial or total renovation or replacement of a compressed air network.
Compressed Air Purification & Piping Monthly e-NewsletterWith a focus on Demand-Side Optimization, compressed air dryers, filters, condensate management, tanks, piping and pneumatic technologies are profiled. How to ensure system reliability, while reducing pressure drop and demand, is explored through System Assessment case studies. |
Qualification of Compressed Air Systems and Distribution
One of the most asked questions when considering compressed air systems is the need to fully qualify all components of compressed air generation and distribution. The first difficulty here is that, in contrast to a process plant (e.g. filling line for aseptic processes), GMP-compliant planning taking into account a "hygienic design" for an air compressor is almost impossible. Even if air compressors are occasionally offered by the manufacturer as "GMP-compliant," with reference to oil-free operation, this generally only refers to the absence of oil cooling and reduction of the lubricated/particle-releasing parts.
It is therefore a recognized GMP practice that the air compressor, including installation, is checked for its technical suitability under the rules of Good Engineering Practice (GEP), that all technically relevant documents are provided, and that successful commissioning is also documented. Against this background, a classic IQ/OQ qualification seems rather inappropriate. This should be listed or defined in the Validation Master Plan (VMP) of the GMP company under the aspect "GMP versus GEP" (6).
This approach was taken up by ISPE in 2014 and commented as follows in the International Society for Pharmaceutical Engineering (ISPE) “Good Practice Guide Process Gases” as follows:
“Gas production normally follows Good Engineering Practices (GEPs). For further information see the ISPE Good Engineering Practice. Gas is not a medicinal product and does not need to be produced following Good Manufacturing Practices.(6)"
GMP-critical, however, is the qualitative preparation of the compressed air, which is used intended not to have any negative impact to the selected specification and for use in the process area relevant to the product. The suitability of all components required for preparation and distribution in the course of the Design-Qualification (DQ), IQ and OQ phases, including a sufficient Performance Qualification (PQ), is a mandatory part of the qualification/validation and is as a GMP consequence subject to the deviation or change management system.
In an analogy to the GMP/GEP approach, the distribution and processing of the compressed air, used exclusively for technical use outside the cleanroom, which means no direct or indirect product contact, can again be regarded as a GEP system and is therefore not subject to the rules of formal qualification.
In the course of qualification, special attention must be paid to the connected process equipment/consumers of air. Here too it must be ensured that potential overpressure or vacuum conditions within the process have no negative influence on the compressed air system to the internal piping of the system due to risk of backflow of contaminants into piping system.
The system boundary for qualification in the compressed air system should be the transfer point to the process system, e.g., the pressure monitoring sensor of a process system is also checked for qualification and should be subjected to regular calibration if necessary.
Quality inspection/Sampling
With the classification of compressed air as a critical medium in pharmaceutical use, a regular check of the compressed air quality in accordance with internal specification is an indispensable part of the validation system: As with pharmaceutical water systems, a sample drawing plan should also be drawn up as part of the PQ for ongoing operation after commissioning.
The number of monitoring points and the frequency of sampling should be determined and planned on the basis of a risk analysis. This can include, based on the application of compressed air, the complexity of the distribution system (e.g. length, branching) or connection to possible contamination risks.
In any case, the sample drawing should be carried out in such a way that it is safe in itself and without the risk of unwanted external contamination. The selected measuring method should be chosen so that the defined specifications can actually be determined in accordance with GMP. The last point in particular is not exactly easy, since for the individual parameters particles, moisture and oil content, as well as a safe sample for airborne germs. This requires a large number of different measuring technologies and sampling methods have to be used.
Given the challenges involved, it’s important to use the appropriate measuring device for monitoring the physical parameters of compressed gas systems. As an example, a device developed by gmp-experts GmbH and SUTO iTEC enables the measurement of all relevant measurement parameters in compressed air monitoring under valid conditions including airborne germs with the addition of an air germ collector.

Shown is the SUTO iTEC’s S600/AirCheck4 compressed air quality analyzer with an additional sampling unit for isokinetic sampling of particles. The isokinetic sampling device is used to sample air carried particles according to ISO 8573 (3) guidelines.
Ensuring the functionality of the sensors is one of the major challenges in dealing with measurement technology relating to the quality inspection of GMP-compliant gases. Here, too, it is an essential GMP requirement the measuring sensors used undergo regular calibration. Hereby it is important to ensure the traceability to the national standard in which all measuring devices have to be traceable to this highest accuracy instance of a physical measured variable. All calibrations carried out have to comply with the requirements of the EU-GMP guidelines and have to be fully recorded in accordance with good documentation practice.
Ensuring Safe Manufacturing
GMP in compressed air systems is the risk-based interaction between the definition of quality parameters and their implementation in a suitable technology for production and distribution. But only GMP-compliant qualification and regular sampling can ensure that the risk of contamination of the pharmaceutical is avoided.
About the Authors
Wolfgang Rudloff, founder and CEO of gmp-experts GmbH, is a mechanical engineer and certified expert in cleanroom technology and GMP management, He has 30 years of management experience in the pharmaceutical industry.
Simon Gleissner is a Product Manager for Air Quality Instruments at SUTO iTEC, email: s.gleissner@SUTO-iTEC.com.
About gmp-experts GmbH
Founded in 2008, gmp-experts GmbH is an internationally operating consulting company for the pharmaceutical, cosmetic and food industries with focus on GMP consulting, project management and GMP training. It specializes in evaluation and holistic improvement of active quality systems, optimization of process and logistics chains, deviation and change management, certified auditing of suppliers and service providers, and assumption of interim mandates. For more information, visit gmp-experts.de.
About SUTO iTEC
SUTO iTEC products play a vital role in applications of leading worldwide companies for the measurement and monitoring of compressed air and gas systems. Since its founding in 2005, SUTO iTEC formerly known as CS-iTEC) has never stopped innovating reliable measurement technology. Besides a few merchandise articles, all SUTO products are developed and manufactured in-house - with a strong focus on quality and customer benefits. For us, service implies being close to our customers, finding individual solutions and reacting quickly when needed. We achieve this service definition through our international presence - with main locations in Germany and China, as well as subsidiaries and long-term partners in 20-plus countries. We are able to bring together the German pursuit of precision and quality with the Asian drive for innovation and speed, four attributes that are essentials for any market leader. For more information, visit www.suto-itec.com.
All photos courtesy of SUTO iTEC.
To read more Pharmaceutical Industry articles, please visit www.airbestpractices.com/industries/pharmaceutical
References
1) EG GMP Guide, Annex 1 "Manufacture of Sterile Drugs," March 2009.
2) EG GMP Guide, Annex 15 "Qualification and Validation," October 2015.
3) ISO 8573-1:2010.
4) ZLG Aide Memoire 07120604 "Monitoring Sterile Manufacturers," January 2015.
5) VDMA standard sheet 15390-1, December 2014.
6) ISPE "Good Practice Guide Process Gases," July 2011.